What is retinoblastoma?
Retinoblastoma is the most common eye cancer for children. It starts from the cells of retina. Retina is a nerve tissue layer that lines the back of the eye. It senses the light and creates a nerve impulse that delivers the information to the brain[1].
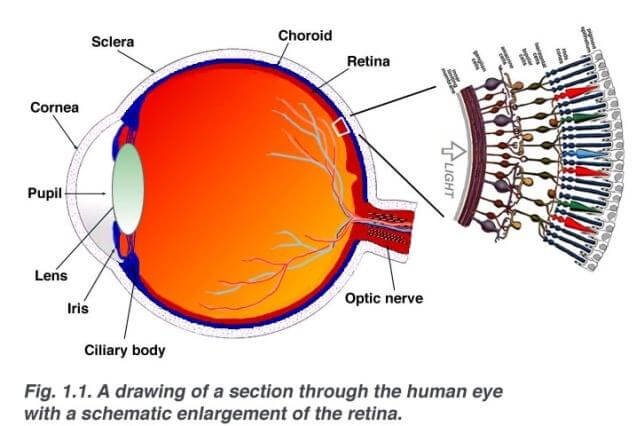
Causes of retinoblastoma
Retinoblastoma is caused by uncontrolled retinal cell division. This process starts early in the fetal development before the cells are matured into normal, mature retinal cells. The uncontrolled cell division is caused by mutation in RB1 gene. RB1 is a tumor suppressor gene that normally prevents cells from uncontrolled division. This gene is also associated with bladder cancer, lung and breast cancer, osteosarcoma and melanoma. There are two types of retinoblastoma:
Congenital retinoblastoma
- Mutation in RB1 gene is congenital
- Changes in the gene occur in the initial stages of fetal development
- Mutation is present in all cells in the body- germline mutation
- Mostly no family history of cancer
- Usually affects both eyes
Because the RB1 gene mutation is present in all the cells in the body, these children often develop other types of cancer. Most commonly affected children develop brain cancer, usually in the pineal gland.
Sporadic retinoblastoma
- Mutation in the RB1 gene develops in only one cell of the eye
- Tumor affects only one eye
- Appears in older age than congenital retinoblastoma
- Affected individuals don’t have a higher risk of any other type of cancer[1,2]
Symptoms of retinoblastoma
Around 5% of affected patients have a positive family history of retinoblastoma.
The most common symptom for retinoblastoma is the cat´s eye reflex (leukocoria)- the pupil appears white when shining light on the eye.
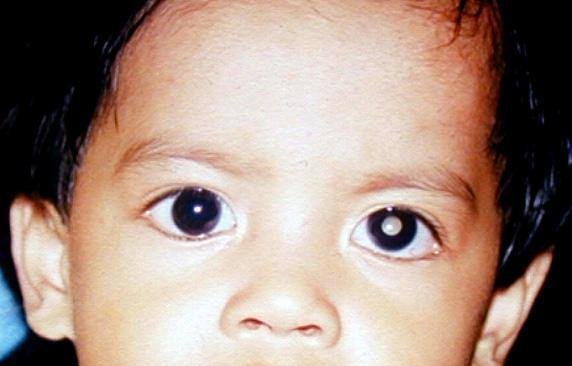
![]()
Other symptoms are:
- Strabismus- improper eye alignment
- Redness, pain in the eye, glaucoma
- Poor vision
- Orbital cellulitis- inflammation of the tissue behind the orbit
- Mydriasis- dilatation of the pupil
- Iris heterochromia
- Hyphema- blood in the front chamber of the eye
- Nystagmus
- White spots in the iris
- Anorexia, trouble gaining weight
- In late stages tissue can exten out of the eye socket (appearance similar to rhabdomyosarcoma in the eye)
Due to necrosis of the tumor, retinoblastoma can also cause secondary changes- inflammation, glaucoma and retinal detachment [3].
 Diagnosis
Diagnosis
After obtaining thorough patient history, eye examination has to be done in order to assess the patient´s vision impairment as well as possible secondary damage caused by retinoblastoma [3].
Laboratory studies
Blood and urine analysis are usually obtained to exclude other conditions. Diagnosis can be made by genetic DNA testing. There are various tests to detect mutation in RB1 gene. Usually DNA testing is also done for parents and siblings in order to assess the possibility of another child being born with retinoblastoma.
Immunohistochemistry can help to determine if the tumor comes from embryological cells or already mature cell. Finding of interphotoreceptor retinoid-binding protein suggests embryonic origin of the tumor.
Bone marrow aspiration and lumbar puncture should be done to investigate further spread of the tumor. This is important, because retinoblastoma first spreads with blood to bone marrow and than through the optic nerve in the cerebrospinal fluid [3].
Histology
Usually the material consists of collections of small blue cells. Frequently necrosis is found in areas distant from vessels and calcification. The most typical sign is Flexner-Wintersteiner rosettes. Also basophilic deposits around blood vessels can be found, as well as cells in mitosis and apoptosis [4].
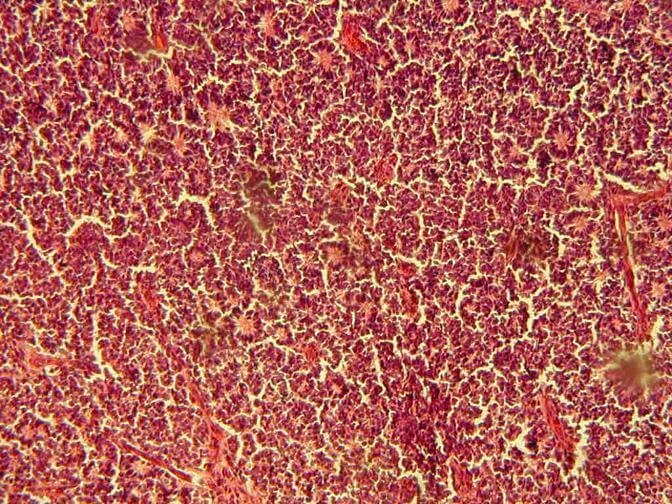
![]()
Imaging studies
- X-ray imaging can detect intraocular calcifications which are a sign of retinoblastoma. It is used only in areas of the world where CT and MRI are not available
- CT- provides a good image for calcifications in the tumor, as well as its extent. It also allows assessing the optic nerve for possible damage.
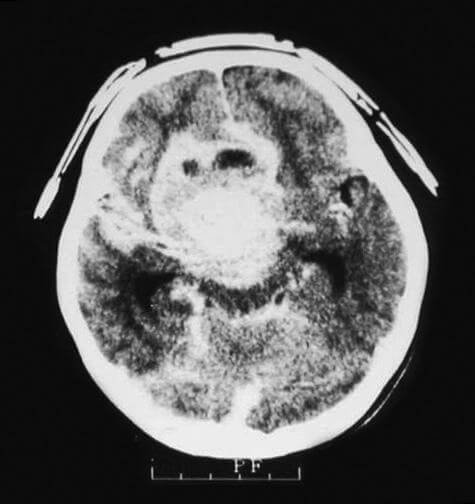
![]()
- MRI- can be helpful in assessing the grade of cell differentiation, but is not as sensitive as CT
- Ultrasound-can help to differentiate retinoblastoma from other non-cancerous formations[3]
Treatment
Radiation therapy
Radiation therapy is effective in locally controlling the tumor. However, there is a high mortality and morbidity rate. Radiation therapy stops the bones from growing therefore patients who have undergone external beam radiation therapy often have facial hypoplasia. It also increases the risk of developing cancer later in life. Radiation therapy is indicated for patients who have:
- Progression of disease during chemotherapy
- Tumors that have extended further than optic nerve
- Significant vitreous seeding- small pieces of the tumor in vitreous corpus
A new method of radiation treatment is using radioactive isotope plaques. A plaque is placed on the eye socket tissue facing the tumor. It is a directed, local treatment of the tumor and minimizes the affect on other tissue. However, it can only be used for smaller sized tumors and it affects sclera tissue [5]. This treatment is also used for uveal melanoma.
Chemotherapy
Chemotherapy is usually used prior to radiation therapy to reduce the size of the tumor. It is also used during radiation therapy to increase the success.
Surgery
Surgical treatment is the standard choice of treatment for cases with poor prognosis. There are various treatment options:
- Enucleation – complete removal of the eye, leaving the orbital tissue intact. Done in patients with complete loss of vision
- Cryotherapy– freezing the tumor tissue. Can be done in tumors that are located anteriorly.
- Photocoagulation – used for posteriorly located tumors. This procedure has a high risk of damaging the optic nerve.
- Exenteration– extension of the tumor in surrounding areas. This procedure is now only done in underdeveloped countries [5]
Prognosis
The prognosis of retinoblastoma depends on many factors, such as age of the patient, extent of the tumor, presence of secondary disease, grade and stage of the tumor. The 5 year survival rate is around 95%. The prognosis is poor if pineal gland tumor is also present. The cure rate is around 90% if enuclation is performed before the tumor spreads to optic nerve. Death usually occurs due to cancer spread in the brain[6].
If you find this article helpful, share it on social media. For your personal experience and other inquiries use the comments box below.
References
- General overview: http://www.cancer.org/cancer/retinoblastoma/detailedguide/retinoblastoma-what-is-retinoblastoma
- Causes: https://ghr.nlm.nih.gov/condition/retinoblastoma#diagnosis
- Symptoms and diagnostics: http://emedicine.medscape.com/article/1222849-clinical#b1
- Histology: http://www.pathologyoutlines.com/topic/eyeretinaretinoblastoma.html
- Treatment: http://www.mayoclinic.org/diseases-conditions/retinoblastoma/diagnosis-treatment/treatment/dxc-20157142
- Prognosis: http://www.cancer.ca/en/cancer-information/cancer-type/retinoblastoma/prognosis-and-survival/?region=on
Similar Posts:
- Tumor Suppressor Genes
- Nerve Sheath Tumor
- Inoperable Brain Tumor
- Acoustic Neuroma – Symptoms, Surgery, Treatment and Prognosis
- Rhabdoid Tumor
- HER2 Positive Breast Cancer
- Medulloblastoma





Leave a Reply